
PCL
PCL
Planar Carbon Lattices
RTG-2861-PCL
C2 - Accurate prediction of multi-electron core excitations in PCLs
Principal investigator: Dr. Dorothea Golze, TU Dresden, [webpage]
Doctoral researcher in the 1st cohort: Jannis Kocklauener
Doctoral researcher in the 2nd cohort: Nargiz Damirova
XPS is a powerful tool to characterize PCLs. An XPS spectrum can show satellite features next to the most important source of chemical information, the photoelectric peak. Satellites are due to multi-electron excitations and are at higher binding energies than the main line. Their intensity is typically very weak for small molecular systems. However, they can have substantial spectral weight for PCLs (Figure), which can complicate the assignment of the photoelectric signals. The analysis of the satellites can greatly complement the information gained from the main excitations. For example, XPS spectra of planar functionalized porphyrin networks show characteristic shake-up satellites depending on the bonding strength with the substrate and the number of layers. However, XPS spectra are generally difficult or even impossible to interpret without aid from theory. Currently, computational XPS is based almost exclusively Kohn-Sham DFT. Although computationally efficient, DFT-based approaches are often not accurate enough to resolve the photoelectric peaks and are - by design - unable to predict satellite features. We will develop accurate computational methods for XPS, which also account for satellites.
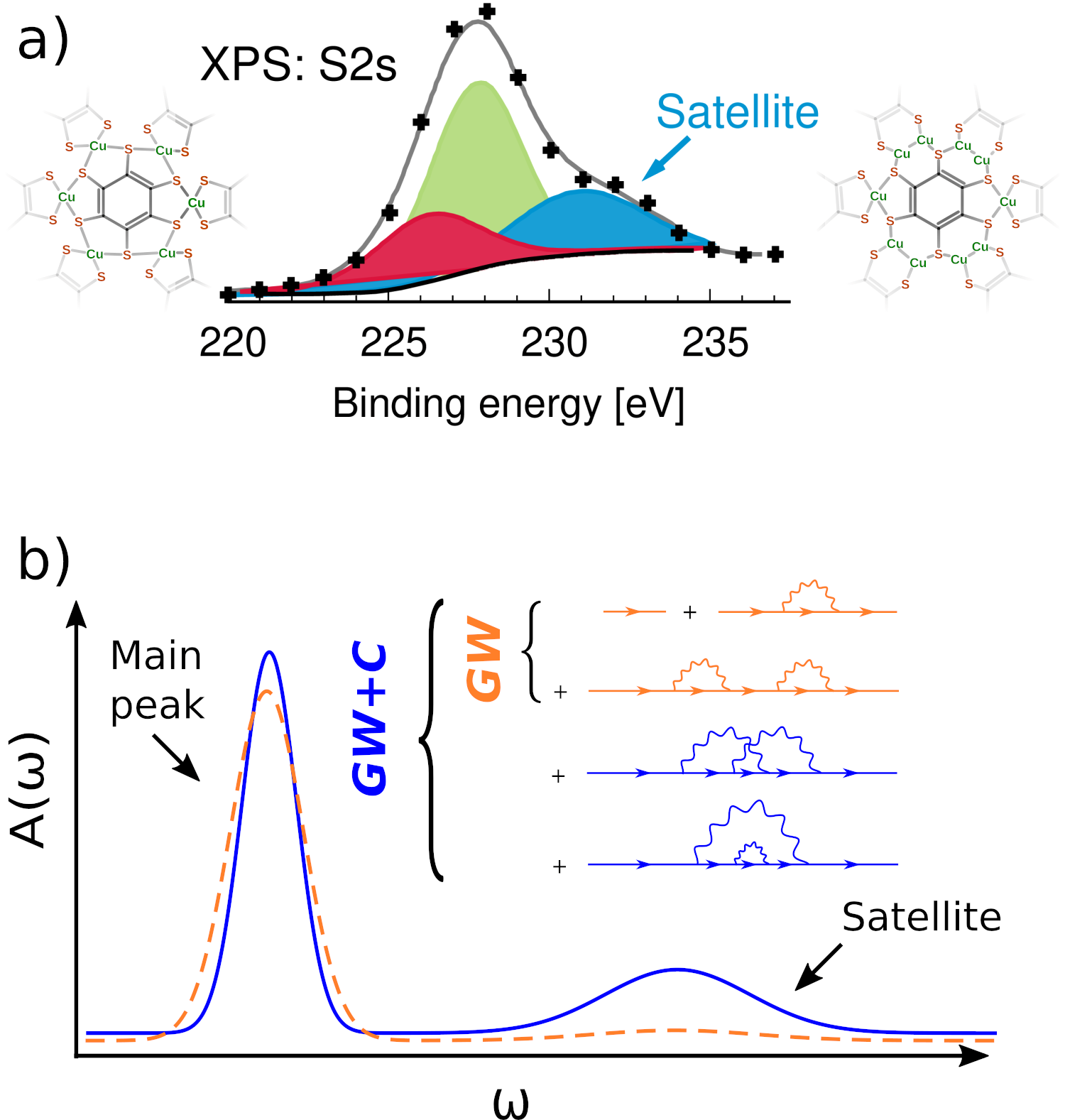
Figure: a) Experimental XPS of a sulfur-containing PCL with two possible structures. b) Comparison of the spectral function A(ω) from GW and GW+C showing that GW+C contains additional Feynman diagrams needed to reproduce satellites.
Thesis topic 1st cohort: Development of the GW+C method for core levels
We recently advanced the highly accurate GW method[1] to deep core-level excitations.[2] While the GW approximation yields accurate photoelectric (main) peaks, the satellite positions are generally not well described (see Figure). We will extend our core-level GW implementation in the program package FHI-aims to the cumulant expansion of the Green’s function (GW+C).[1] The GW+C method includes so-called vertex corrections beyond GW and was already successfully applied for the computation of plasmon satellites for valence shell excitations.[3] We will investigate the suitability of the cumulant expansion for deep core states, starting with small molecules like H2O and CO, for which high-resolution reference experimental data are available.
Thesis topic 2nd cohort: Extension of the GW+C approach to materials
[2] D. Golze, L. Keller, P. Rinke, “Accurate absolute and relative core-level binding energies from GW”, J. Phys. Chem. Lett. 2020, 11, 1840.
[3] J. S. Zhou, J. J. Kas, L. Sponza, I. Reshetnyak, M. Guzzo, C. Giorgetti, M. Gatti, F. Sottile, J. J. Rehr, L. Reining, J. Chem. Phys. 2015, 143, 184109.